
미국 식품의약국(FDA, U.S. Food and Drug Administration)은 국민 보건을 책임지는 핵심 규제 기관으로, 의약품, 의료기기, 식품, 화장품 등 다양한 제품의 안전성과 효과를 철저히 관리합니다. 특히 신약 개발 과정에서 FDA의 역할과 규제 절차를 이해하는 것은 글로벌 제약 산업에서 성공하기 위한 필수 요소입니다.
미국 FDA의 구조와 역할
FDA는 보건복지부(Department of Health and Human Services, HHS) 산하에 있으며, 그 구조는 여러 센터와 사무국으로 나뉘어 각기 다른 규제 영역을 담당합니다. 신약 허가와 관련하여 중요한 역할을 하는 주요 센터는 다음과 같습니다:
ㆍCDER(Center for Drug Evaluation and Research): 화학 합성 의약품 및 생물의약품 일부의 안전성, 효과 및 품질을 평가하여 시판을 승인합니다. 대부분의 신약 허가 심사를 담당합니다.
ㆍCBER(Center for Biologics Evaluation and Research): 백신, 혈액 제품, 유전자 치료제, 세포 치료제 등 생물학적 제제의 안전성과 효과를 규제합니다.
ㆍCDRH(Center for Devices and Radiological Health): 의료기기와 방사선 방출 제품을 규제합니다.
ㆍCFSAN(Center for Food Safety and Applied Nutrition): 식품과 화장품의 안전성을 관리합니다.
ㆍOC(Office of the Commissioner) 및 ORA(Office of Regulatory Affairs): ORA는 현장 검사(Inspection) 및 집행 활동을 수행하는 FDA의 주요 조직으로, 제조소 실사 등을 담당합니다.
이러한 조직을 통해 FDA는 신약이 대중에게 안전하고 효과적이며 품질이 보장되는 방식으로 생산될 수 있도록 엄격하게 관리합니다.
신약 허가 절차 중 핵심 단계: 실사(Inspection)
신약 허가 절차는 크게 비임상시험(Pre-clinical), 임상시험(Clinical Trials), 그리고 최종적으로 신약 허가 신청(NDA/BLA) 및 심사 단계를 거칩니다. 이 중 심사 단계의 후반부에 큰 변화를 초래할 수 있는 절차로 실사(Inspection) 단계가 있습니다.
실사는 단순히 제조 시설만 확인하는 것이 아니라, 임상시험 데이터의 신뢰성까지 아우르는 광범위한 규제 준수 여부 검증 활동입니다. FDA의 실사는 크게 신약의 제조 과정을 다루는 GMP 실사와 임상 시험 과정을 다루는 GCP 실사로 나뉩니다.
◆GMP(Good Manufacturing Practice) 실사
GMP 실사는 의약품의 제조, 가공, 포장, 보관 등에 관한 품질 시스템이 FDA의 우수 제조 관리 기준 규정을 충족하는지 판단합니다. 신약이 신청서에 명시된 기준과 GMP를 준수하여 일관성 있게 생산될 수 있는지 확인합니다.
실사 대상은 의약품 제조 시설 및 품질 관리 시스템(Quality Management System, QMS)이며, 신약의 안전성과 유효성에 직접적인 영향을 미치는 제품 품질을 보장하는 핵심이라 할 수 있습니다. 특히 승인 전 실사(Pre-Approval Inspection, PAI)는 신약 허가를 받기 위한 필수 관문입니다.
◆GCP(Good Clinical Practice) 실사
GCP 실사는 임상 시험 데이터의 신뢰성과 임상 참여자의 안전 및 권리가 보호되었는지 확인하기 위해 우수 임상 관리 기준 준수 여부를 평가합니다. 이를 BIMO(Bioresearch Monitoring) 프로그램의 일환으로 수행합니다.
실사 대상은 임상 시험을 수행한 병원/기관(Investigational Sites), 임상 시험을 관리한 스폰서(Sponsor), 임상시험 수탁기관(CRO)이며, 임상 시험 과정에서 수집된 데이터의 무결성(Data Integrity)을 검증하고, 시험기관, 임상의, 스폰서가 GCP 규정을 준수했는지 확인합니다.
임상 시험 데이터는 신약의 유효성과 안전성을 입증하는 유일한 근거이므로, GCP 실사는 이 데이터의 신뢰성을 확보하는 데 결정적입니다.
실사 결과의 분류
실사 결과는 규정 위반의 경중에 따라 다음 세 가지 등급으로 분류됩니다:
ㆍNAI(No Action Indicated): 어떠한 규제 조치도 필요하지 않음.
ㆍVAI(Voluntary Action Indicated): 사소한 위반 사항이 발견되었으나, 회사가 자발적으로 시정할 것을 권고.
ㆍOAI(Official Action Indicated): 중대한 위반 사항이 발견되어 공식적인 규제 조치가 필요함. 이 등급은 종종 경고 서한인 'Warning Letter'로 이어집니다.
현장 지적 사항을 담은 문서: FDA Form 483

FDA 조사관은 실사 과정에서 GMP, GCP, GLP 등 관련 법규의 위반 소지가 있거나 결함이 있다고 판단되는 조건이나 관찰 사항을 발견하면, 실사 종료 시 해당 시설/기관의 경영진에게 'FDA Form 483'(Inspectional Observations)을 발행합니다.
◆Form 483의 특징
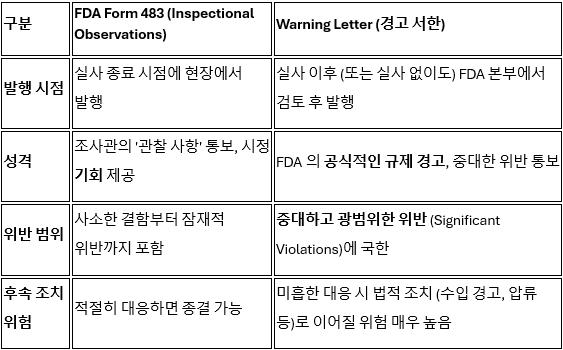
ㆍ발행 시점: 실사 종료 회의(Close-out meeting) 시점에 발행됩니다.
ㆍ내용: 조사관이 발견한 구체적인 관찰 사항(observations)을 기록합니다. 이는 품질 시스템의 취약점이나 잠재적인 규정 위반을 나타냅니다.
ㆍ대응 의무: Form 483 자체는 공식적인 법적 조치는 아니지만, 회사가 FDA 규정을 준수하지 못하고 있음을 공식적으로 통보하는 문서입니다.
ㆍ회사의 대응: Form 483을 수령한 회사는 일반적으로 15 영업일 이내에 FDA에 공식적인 대응 계획을 제출해야 합니다. 이 대응 계획은 지적된 각 관찰 사항에 대한 시정 조치(Corrective and Preventive Actions, CAPA)와 그 이행 일정을 상세히 포함해야 합니다.
핵심은 Form 483이 '시정 권고'의 성격을 가지며, 회사가 신속하고 철저하게 대응할 수 있는 기회를 제공한다는 점입니다. 만약 회사의 대응이 부적절하거나, 지적 사항이 매우 중대하여 OAI 등급으로 분류되면, 다음 단계인 Warning Letter로 격상될 가능성이 높아집니다.
공식적인 규제 경고: 경고 서한(Warning Letter)

Warning Letter는 Form 483보다 훨씬 더 심각하고 공식적인 규제 조치(enforcement action)의 시작을 알리는 문서입니다. 이는 Form 483을 통해 지적된 위반 사항을 회사가 적절하게 시정하지 않았거나, 실사에서 발견된 위반 사항 자체가 매우 중대하고 광범위하여 즉각적인 공식 조치가 필요하다고 FDA가 판단했을 때 발행됩니다.
◆Warning Letter의 중대성
발행 사유: 주로 중대한 규정 위반(Significant Violations)에 대해 발행되며, 특히 환자 안전이나 제품 품질에 직접적인 영향을 미칠 수 있는 중대한 GMP 또는 GCP 위반인 경우에 해당됩니다.
법적 경고: Warning Letter는 위반 사항이 시정되지 않을 경우 FDA가 제품 압류, 수입 금지(Import Alert), 강제 리콜, 형사 기소와 같은 추가적인 법적 조치를 취할 수 있음을 공식적으로 경고하는 문서입니다.
공개: 대부분의 Warning Letter는 FDA 웹사이트에 공개되어 해당 기업의 신뢰도와 평판에 심각한 타격을 줍니다.
회사의 대응: Warning Letter를 받은 회사는 일반적으로 15영업일 이내에 위반 사항을 시정하고 향후 재발 방지를 위한 종합적이고 철저한 CAPA 계획 및 이행 증거를 FDA에 제출해야 합니다. 단순히 문제 해결을 넘어 근본 원인 분석(Root Cause Analysis)을 통한 시스템 개선이 필수적입니다.
Form 483과 Warning Letter의 차이
미국 FDA의 실사 과정에서 발행되는 Form 483과 Warning Letter는 단순한 규제 문서가 아니라, 제약·바이오 기업이 반드시 인식해야 할 핵심적인 규제 리스크이자, 동시에 기업의 품질(Quality) 및 규정 준수(Compliance) 전략을 점검하는 중요한 전환점입니다.
Form 483은 현장의 구체적인 관찰 사항(Observations)을 통해 잠재적 결함을 조기에 인지하고 시정할 수 있는 기회를 제공합니다. 반면, Warning Letter는 중대한 규정 위반에 대한 공식적인 집행 조치(Enforcement Action)의 시작을 알리며, 미조치 시 시장 접근 제한(Import Alert)이나 법적 제재로 이어지는 심각한 리스크입니다.
규제 준수와 신뢰의 중요성
미국 FDA 실사 성공의 핵심은 '실사를 위한 준비'가 아닌, '항상 실사를 받을 수 있는 상태를 유지'하는 데 달려 있습니다. 이는 철저한 데이터 무결성(Data Integrity) 관리, 선제적인 자체 감사(Self-Inspection), 그리고 근본 원인 분석에 기반한 효율적인 CAPA 시스템 운영을 통해 가능합니다. 이러한 일상적인 노력이 곧 지속 가능한 고품질 운영을 보장하고, 기업의 글로벌 경쟁력과 신뢰도를 확보하는 가장 강력한 규제 대비책이 될 것입니다.